


This HTML version of
You might prefer to read the PDF version, or you can buy a hard copy from Amazon.
Chapter 13 Simulation
In this chapter I describe my solution to a problem posed by a patient with a kidney tumor. I think the problem is important and relevant to patients with these tumors and doctors treating them.
And I think the solution is interesting because, although it is a Bayesian approach to the problem, the use of Bayes’s theorem is implicit. I present the solution and my code; at the end of the chapter I will explain the Bayesian part.
If you want more technical detail than I present here, you can read my paper on this work at http://arxiv.org/abs/1203.6890.
13.1 The Kidney Tumor problem
I am a frequent reader and occasional contributor to the online statistics forum at http://reddit.com/r/statistics. In November 2011, I read the following message:
"I have Stage IV Kidney Cancer and am trying to determine if the cancer formed before I retired from the military. ... Given the dates of retirement and detection is it possible to determine when there was a 50/50 chance that I developed the disease? Is it possible to determine the probability on the retirement date? My tumor was 15.5 cm x 15 cm at detection. Grade II."
I contacted the author of the message and got more information; I learned that veterans get different benefits if it is "more likely than not" that a tumor formed while they were in military service (among other considerations).
Because renal tumors grow slowly, and often do not cause symptoms, they are sometimes left untreated. As a result, doctors can observe the rate of growth for untreated tumors by comparing scans from the same patient at different times. Several papers have reported these growth rates.
I collected data from a paper by Zhang et al1. I contacted the authors to see if I could get raw data, but they refused on grounds of medical privacy. Nevertheless, I was able to extract the data I needed by printing one of their graphs and measuring it with a ruler.
Figure 13.1: CDF of RDT in doublings per year.
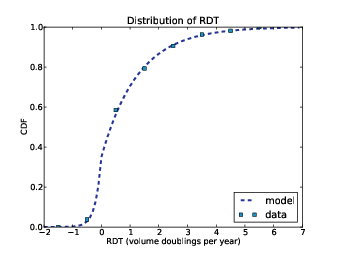
They report growth rates in reciprocal doubling time (RDT), which is in units of doublings per year. So a tumor with RDT=1 doubles in volume each year; with RDT=2 it quadruples in the same time, and with RDT=−1, it halves. Figure 13.1 shows the distribution of RDT for 53 patients.
The squares are the data points from the paper; the line is a model I fit to the data. The positive tail fits an exponential distribution well, so I used a mixture of two exponentials.
13.2 A simple model
It is usually a good idea to start with a simple model before trying something more challenging. Sometimes the simple model is sufficient for the problem at hand, and if not, you can use it to validate the more complex model.
For my simple model, I assume that tumors grow with a constant doubling time, and that they are three-dimensional in the sense that if the maximum linear measurement doubles, the volume is multiplied by eight.
I learned from my correspondent that the time between his discharge from the military and his diagnosis was 3291 days (about 9 years). So my first calculation was, “If this tumor grew at the median rate, how big would it have been at the date of discharge?”
The median volume doubling time reported by Zhang et al is 811 days. Assuming 3-dimensional geometry, the doubling time for a linear measure is three times longer.
# time between discharge and diagnosis, in days
interval = 3291.0
# doubling time in linear measure is doubling time in volume * 3
dt = 811.0 * 3
# number of doublings since discharge
doublings = interval / dt
# how big was the tumor at time of discharge (diameter in cm)
d1 = 15.5
d0 = d1 / 2.0 ** doublings
You can download the code in this chapter from http://thinkbayes.com/kidney.py. For more information see Section 0.3.
The result, d0, is about 6 cm. So if this tumor formed after the date of discharge, it must have grown substantially faster than the median rate. Therefore I concluded that it is “more likely than not” that this tumor formed before the date of discharge.
In addition, I computed the growth rate that would be implied if this tumor had formed after the date of discharge. If we assume an initial size of 0.1 cm, we can compute the number of doublings to get to a final size of 15.5 cm:
# assume an initial linear measure of 0.1 cm
d0 = 0.1
d1 = 15.5
# how many doublings would it take to get from d0 to d1
doublings = log2(d1 / d0)
# what linear doubling time does that imply?
dt = interval / doublings
# compute the volumetric doubling time and RDT
vdt = dt / 3
rdt = 365 / vdt
dt is linear doubling time, so vdt is volumetric doubling time, and rdt is reciprocal doubling time.
The number of doublings, in linear measure, is 7.3, which implies an RDT of 2.4. In the data from Zhang et al, only 20% of tumors grew this fast during a period of observation. So again, I concluded that is “more likely than not” that the tumor formed prior to the date of discharge.
These calculations are sufficient to answer the question as posed, and on behalf of my correspondent, I wrote a letter explaining my conclusions to the Veterans’ Benefit Administration.
Later I told a friend, who is an oncologist, about my results. He was surprised by the growth rates observed by Zhang et al, and by what they imply about the ages of these tumors. He suggested that the results might be interesting to researchers and doctors.
But in order to make them useful, I wanted a more general model of the relationship between age and size.
13.3 A more general model
Given the size of a tumor at time of diagnosis, it would be most useful to know the probability that the tumor formed before any given date; in other words, the distribution of ages.
To find it, I run simulations of tumor growth to get the distribution of size conditioned on age. Then we can use a Bayesian approach to get the distribution of age conditioned on size.
The simulation starts with a small tumor and runs these steps:
- Choose a growth rate from the distribution of RDT.
- Compute the size of the tumor at the end of an interval.
- Record the size of the tumor at each interval.
- Repeat until the tumor exceeds the maximum relevant size.
For the initial size I chose 0.3 cm, because carcinomas smaller than that are less likely to be invasive and less likely to have the blood supply needed for rapid growth (see http://en.wikipedia.org/wiki/Carcinoma_in_situ).
I chose an interval of 245 days (about 8 months) because that is the median time between measurements in the data source.
For the maximum size I chose 20 cm. In the data source, the range of observed sizes is 1.0 to 12.0 cm, so we are extrapolating beyond the observed range at each end, but not by far, and not in a way likely to have a strong effect on the results.
Figure 13.2: Simulations of tumor growth, size vs. time.
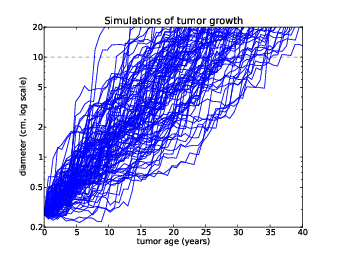
The simulation is based on one big simplification: the growth rate is chosen independently during each interval, so it does not depend on age, size, or growth rate during previous intervals.
In Section 13.7 I review these assumptions and consider more detailed models. But first let’s look at some examples.
Figure 13.2 shows the size of simulated tumors as a function of age. The dashed line at 10 cm shows the range of ages for tumors at that size: the fastest-growing tumor gets there in 8 years; the slowest takes more than 35.
I am presenting results in terms of linear measurements, but the calculations are in terms of volume. To convert from one to the other, again, I use the volume of a sphere with the given diameter.
13.4 Implementation
Here is the kernel of the simulation:
def MakeSequence(rdt_seq, v0=0.01, interval=0.67, vmax=Volume(20.0)):
seq = v0,
age = 0
for rdt in rdt_seq:
age += interval
final, seq = ExtendSequence(age, seq, rdt, interval)
if final > vmax:
break
return seq
rdt_seq is an iterator that yields
random values from the CDF of growth rate.
v0 is the initial volume in mL. interval is the time step
in years. vmax is the final volume corresponding to a linear
measurement of 20 cm.
Volume converts from linear measurement in cm to volume in mL, based on the simplification that the tumor is a sphere:
def Volume(diameter, factor=4*math.pi/3):
return factor * (diameter/2.0)**3
ExtendSequence computes the volume of the tumor at the end of the interval.
def ExtendSequence(age, seq, rdt, interval):
initial = seq[-1]
doublings = rdt * interval
final = initial * 2**doublings
new_seq = seq + (final,)
cache.Add(age, new_seq, rdt)
return final, new_seq
age is the age of the tumor at the end of the interval. seq is a tuple that contains the volumes so far. rdt is the growth rate during the interval, in doublings per year. interval is the size of the time step in years.
The return values are final, the volume of the
tumor at the end of the interval, and new_seq, a new
tuple containing the volumes in seq plus the new volume
final.
Cache.Add records the age and size of each tumor at the end of each interval, as explained in the next section.
13.5 Caching the joint distribution
Figure 13.3: Joint distribution of age and tumor size.
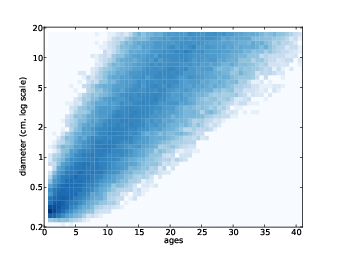
Here’s how the cache works.
class Cache(object):
def __init__(self):
self.joint = thinkbayes.Joint()
joint is a joint Pmf that records the frequency of each age-size pair, so it approximates the joint distribution of age and size.
At the end of each simulated interval, ExtendSequence calls Add:
# class Cache
def Add(self, age, seq):
final = seq[-1]
cm = Diameter(final)
bucket = round(CmToBucket(cm))
self.joint.Incr((age, bucket))
Again, age is the age of the tumor, and seq is the sequence of volumes so far.
Figure 13.4: Distributions of age, conditioned on size.
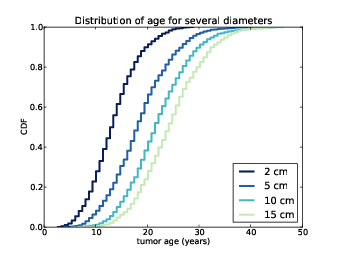
Before adding the new data to the joint distribution, we use Diameter to convert from volume to diameter in centimeters:
def Diameter(volume, factor=3/math.pi/4, exp=1/3.0):
return 2 * (factor * volume) ** exp
And CmToBucket to convert from centimeters to a discrete bucket number:
def CmToBucket(x, factor=10):
return factor * math.log(x)
The buckets are equally spaced on a log scale. Using factor=10 yields a reasonable number of buckets; for example, 1 cm maps to bucket 0 and 10 cm maps to bucket 23.
After running the simulations, we can plot the joint distribution as a pseudocolor plot, where each cell represents the number of tumors observed at a given size-age pair. Figure 13.3 shows the joint distribution after 1000 simulations.
13.6 Conditional distributions
Figure 13.5: Percentiles of tumor age as a function of size.
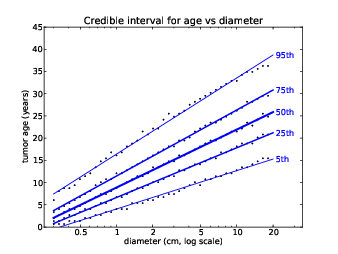
By taking a vertical slice from the joint distribution, we can get the distribution of sizes for any given age. By taking a horizontal slice, we can get the distribution of ages conditioned on size.
Here’s the code that reads the joint distribution and builds the conditional distribution for a given size.
# class Cache
def ConditionalCdf(self, bucket):
pmf = self.joint.Conditional(0, 1, bucket)
cdf = pmf.MakeCdf()
return cdf
bucket is the integer bucket number corresponding to
tumor size. Joint.Conditional computes the
PMF of age conditioned on bucket.
The result is the CDF of age conditioned on bucket.
Figure 13.4 shows several of these CDFs, for a range of sizes. To summarize these distributions, we can compute percentiles as a function of size.
percentiles = [95, 75, 50, 25, 5]
for bucket in cache.GetBuckets():
cdf = ConditionalCdf(bucket)
ps = [cdf.Percentile(p) for p in percentiles]
Figure 13.5 shows these percentiles for each size bucket. The data points are computed from the estimated joint distribution. In the model, size and time are discrete, which contributes numerical errors, so I also show a least squares fit for each sequence of percentiles.
13.7 Serial Correlation
The results so far are based on a number of modeling decisions; let’s review them and consider which ones are the most likely sources of error:
- To convert from linear measure to volume, we assume that tumors are approximately spherical. This assumption is probably fine for tumors up to a few centimeters, but not for very large tumors.
- The distribution of growth rates in the simulations are based on a continuous model we chose to fit the data reported by Zhang et al, which is based on 53 patients. The fit is only approximate and, more importantly, a larger sample would yield a different distribution.
- The growth model does not take into account tumor subtype or grade; this assumption is consistent with the conclusion of Zhang et al: “Growth rates in renal tumors of different sizes, subtypes and grades represent a wide range and overlap substantially.” But with a larger sample, a difference might become apparent.
- The distribution of growth rate does not depend on the size of
the tumor. This assumption would not be realistic for very
small and very large tumors, whose growth is limited by blood supply.
But tumors observed by Zhang et al ranged from 1 to 12 cm, and they found no statistically significant relationship between size and growth rate. So if there is a relationship, it is likely to be weak, at least in this size range.
- In the simulations, growth rate during each interval is independent of previous growth rates. In reality it is plausible that tumors that have grown quickly in the past are more likely to grow quickly. In other words, there is probably a serial correlation in growth rate.
Of these, the first and last seem the most problematic. I’ll investigate serial correlation first, then come back to spherical geometry.
To simulate correlated growth, I wrote a generator2 that yields a correlated series from a given Cdf. Here’s how the algorithm works:
- Generate correlated values from a Gaussian distribution. This is easy to do because we can compute the distribution of the next value conditioned on the previous value.
- Transform each value to its cumulative probability using the Gaussian CDF.
- Transform each cumulative probability to the corresponding value using the given Cdf.
Here’s what that looks like in code:
def CorrelatedGenerator(cdf, rho):
x = random.gauss(0, 1)
yield Transform(x)
sigma = math.sqrt(1 - rho**2);
while True:
x = random.gauss(x * rho, sigma)
yield Transform(x)
cdf is the desired Cdf; rho is the desired correlation. The values of x are Gaussian; Transform converts them to the desired distribution.
The first value of x is Gaussian with mean 0 and standard deviation 1. For subsequent values, the mean and standard deviation depend on the previous value. Given the previous x, the mean of the next value is x * rho, and the variance is 1 - rho**2.
Transform maps from each Gaussian value, x, to a value from the given Cdf, y.
def Transform(x):
p = thinkbayes.GaussianCdf(x)
y = cdf.Value(p)
return y
GaussianCdf computes the CDF of the standard Gaussian distribution at x, returning a cumulative probability. Cdf.Value maps from a cumulative probability to the corresponding value in cdf.
Depending on the shape of cdf, information can be lost in transformation, so the actual correlation might be lower than rho. For example, when I generate 10000 values from the distribution of growth rates with rho=0.4, the actual correlation is 0.37. But since we are guessing at the right correlation anyway, that’s close enough.
Remember that MakeSequence takes an iterator as an argument. That interface allows it to work with different generators:
iterator = UncorrelatedGenerator(cdf)
seq1 = MakeSequence(iterator)
iterator = CorrelatedGenerator(cdf, rho)
seq2 = MakeSequence(iterator)
In this example, seq1 and seq2 are drawn from the same distribution, but the values in seq1 are uncorrelated and the values in seq2 are correlated with a coefficient of approximately rho.
Now we can see what effect serial correlation has on the results; the following table shows percentiles of age for a 6 cm tumor, using the uncorrelated generator and a correlated generator with target ρ = 0.4.
Serial Diameter Percentiles of age Correlation (cm) 5th 25th 50th 75th 95th 0.0 6.0 10.7 15.4 19.5 23.5 30.2 0.4 6.0 9.4 15.4 20.8 26.2 36.9
Table 13.1: Percentiles of tumor age conditioned on size.
Correlation makes the fastest growing tumors faster and the slowest slower, so the range of ages is wider. The difference is modest for low percentiles, but for the 95th percentile it is more than 6 years. To compute these percentiles precisely, we would need a better estimate of the actual serial correlation.
However, this model is sufficient to answer the question we started with: given a tumor with a linear dimension of 15.5 cm, what is the probability that it formed more than 8 years ago?
Here’s the code:
# class Cache
def ProbOlder(self, cm, age):
bucket = CmToBucket(cm)
cdf = self.ConditionalCdf(bucket)
p = cdf.Prob(age)
return 1-p
cm is the size of the tumor; age is the age threshold in years. ProbOlder converts size to a bucket number, gets the Cdf of age conditioned on bucket, and computes the probability that age exceeds the given value.
With no serial correlation, the probability that a 15.5 cm tumor is older than 8 years is 0.999, or almost certain. With correlation 0.4, faster-growing tumors are more likely, but the probability is still 0.995. Even with correlation 0.8, the probability is 0.978.
Another likely source of error is the assumption that tumors are approximately spherical. For a tumor with linear dimensions 15.5 x 15 cm, this assumption is probably not valid. If, as seems likely, a tumor this size is relatively flat, it might have the same volume as a 6 cm sphere. With this smaller volume and correlation 0.8, the probability of age greater than 8 is still 95%.
So even taking into account modeling errors, it is unlikely that such a large tumor could have formed less than 8 years prior to the date of diagnosis.
13.8 Discussion
Well, we got through a whole chapter without using Bayes’s theorem or the Suite class that encapsulates Bayesian updates. What happened?
One way to think about Bayes’s theorem is as an algorithm for inverting conditional probabilities. Given p(B|A), we can compute p(A|B), provided we know p(A) and p(B). Of course this algorithm is only useful if, for some reason, it is easier to compute p(B|A) than p(A|B).
In this example, it is. By running simulations, we can estimate the distribution of size conditioned on age, or p(size|age). But it is harder to get the distribution of age conditioned on size, or p(age|size). So this seems like a perfect opportunity to use Bayes’s theorem.
The reason I didn’t is computational efficiency. To estimate p(size|age) for any given size, you have to run a lot of simulations. Along the way, you end up computing p(size|age) for a lot of sizes. In fact, you end up computing the entire joint distribution of size and age, p(size, age).
And once you have the joint distribution, you don’t really need Bayes’s theorem, you can extract p(age|size) by taking slices from the joint distribution, as demonstrated in ConditionalCdf.
So we side-stepped Bayes, but he was with us in spirit.
- 1
- Zhang et al, Distribution of Renal Tumor Growth Rates Determined by Using Serial Volumetric CT Measurements, January 2009 Radiology, 250, 137-144.
- 2
- If you are not familiar with Python generators, see http://wiki.python.org/moin/Generators.



